Bästa EFFECTS-vänner
Idén till EFFECTS föddes för snart 7 år sedan. Det var efter resultatet från FLAME-studien som presenterades i Lancet Neurology 2011. Ursprungligen var tanken att genomföra en stor multinationell studie i Storbritannien, Australien, Nya Zeeland och Sverige. Men av praktiska skäl valde vi att starta tre parallella studier med en gemensam design. Frågeställningarna för alla studierna var att undersöka om fluoxetin förbättrar återhämtningen efter stroke.
Resultatet från den i Storbritannien genomförda Fluoxetine Or Control Under Supervision (FOCUS) studien har nu presenterats vid UK Stroke Forum och samtidigt publicerats (Lancet, 5 december 2018).
Det var ingen skillnad mellan de som erhöll fluoxetin eller placebo i någon av de olika mRS-grupperna (common odds ratio, 0,951; 95 % CI: 0,839 till 1,079; p=0,439).
FOCUS randomiserade 3 127 patienter med en akut stroke inom 2 till15 dagar till antingen fluoxetin 20 mg/dag (n=1 564) eller placebo (n=1 563) under 6 månader. Det primära utfallsmåttet, tillgänglig i 99,3 procent i vardera gruppen, var den modifierade Rankin-skalan (mRS) vid 6 månader.
Bland de sekundära utfallsmåtten kunde man se att fluoxetin minskade andelen individer med depression vid sex månader jämfört med placebo (13,0 % fluoxetin vs 16,9% placebo, absolut skillnad 3,9, 95 % CI skillnad 1,26 till 6,30 %; p=0,0033). Men samtidigt var det fler som drabbades av benfrakturer (2,9 % fluoxetin vs 1,5 % placebo, absolut skillnad 1,41 %, 95 CI skillnad 0,38 till 2,43; p=0,0070).
Resultatet visar således att 20 mg fluoxetin/dag under sex månader efter en akut stroke inte påverkar funktionsnivån men att det minskar förekomsten av depression och ökar risken för benfrakturer.
Bedömning
- Utfallet av det primära utfallsmåttet i FOCUS var neutralt, dvs behandling med fluoxetin påverkade inte signifikant funktionsnivån i vare sig positiv eller negativ riktning.
- En ökad förekomst av benfrakturer sågs. Det är av värde att fyndet studeras i andra pågående studier för att se om det kan verifieras.
- Inga andra säkerhetsrisker (såsom ökad blödningsrisk) med behandlingen observerades.
- Resultaten från FOCUS är inte säkert överförbara till andra länder.
- Effekt av fluoxetin på funktion efter stroke kan till exempel interagera med frekvens och duration av sjukgymnastiska behandlingar och med andra rehabiliteringsinsatser, och här skiljer sig Sverige från Storbritannien. I EFFECTS planeras att specifikt ta hänsyn till rehabiliteringsinsatser och fysisk aktivitet i kombination med fluoxetinbehandling. Sverige har bra data jämfört med Storbritannien avseende den fysiska aktiviteten.
- I FOCUS var patienten en aning sjukare (NIHSS medianvärde 6) jämfört med EFFECTS (medianvärde NIHSS = 3).
- Resultat från pågående studier och planerade analyser på individualnivå är viktiga för att viktiga för att få en så fullständig och säker bild som möjligt hur fluoxetin (SSRI) ska användas vid stroke. Finns det subgrupper och markörer för när fluoxetin är till mer nytta?
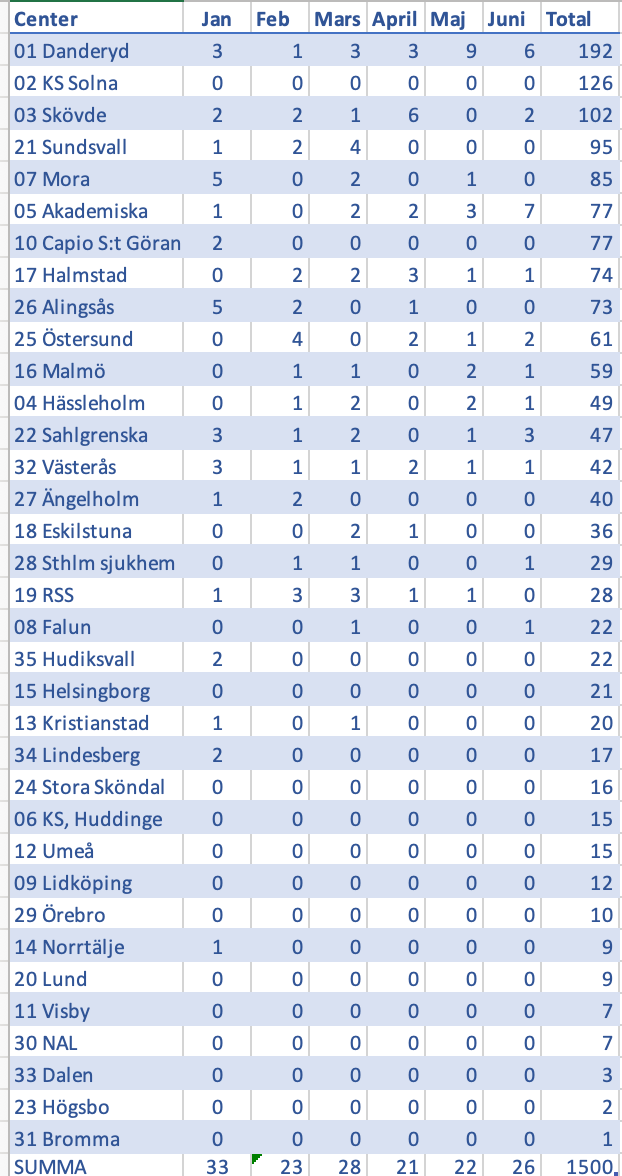
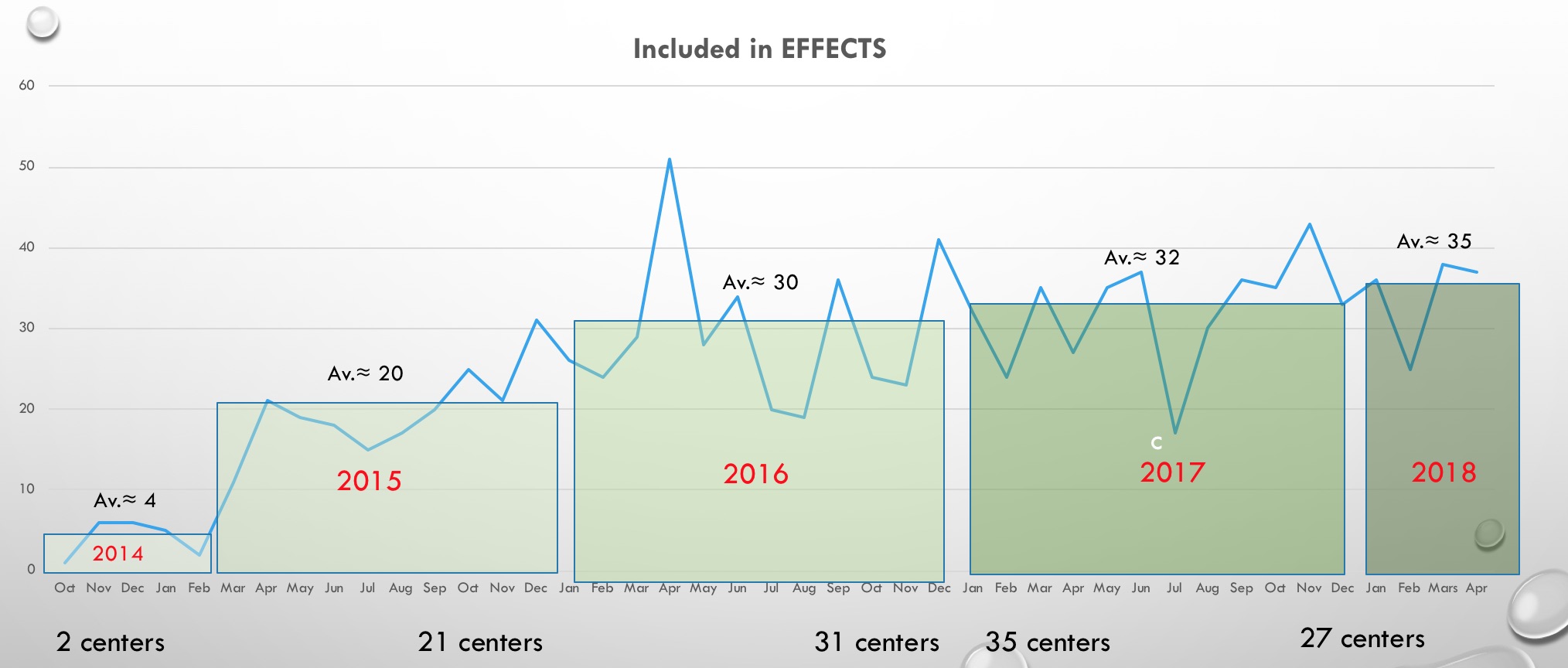
- Styrkommittén bedömer att FOCUS inte visar effekter på primära utfallsmåttet eller säkerhet som gör att det finns skäl att modifiera planeringen för EFFECTS. Vår studie är power-beräknad för 1500 patienter och för att få ett så robust vetenskapligt resultat som möjligt bedömer vi att den planerade studiestorlek inte bör minskas om det inte finns starka skäl från andra studier av visad effekt eller säkerhetsproblem. Ett förtida studieavbrott kan medföra att allt det arbete som lagts ner i EFFECTS så här långt är förgäves. Ansvariga för AFFINITY har gjort motsvarande bedömning liksom att man i FOCUS-publikationen pekar på att det är viktigt att de andra studierna fortsätter.
EFFECTS säkerhetskommitté kommer också att sammanträda för att göra en oberoende bedömning. Deras rapport och rekommendation kommer att publiceras på EFFECTS hemsida och via veckobrev så fort den blir känd av EFFECTS Chief Investigator.
Sammanfattningsvis rekommenderar styrkommittén i nuläget att EFFECTS slutförs som planerat, och vi bedömer det angeläget att den nuvarande goda inklusionstakten kan fortgå oförändrad till det att 1500 inkluderade patienter har uppnåtts.
7 december 2018
Katharina Stibrant Sunnerhagen (ordf), Bo Norrving, Per Wester, Håkan Wallén, Jörgen Borg, Björn Mårtensson, Per Näsman, Eva Isaksson och Erik Lundström för EFFECTS styrkommitté
REFERENS
1) https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(18)32823-X/fulltext